รามา กระตุ้นคนรู้จักโรค “โกเช่ร์” โรคหายากแต่มีอยู่จริง

ศ.พญ. ดวงฤดี วัฒนศิริชัยกุล หัวหน้าสาขาเวชพันธุศาสตร์ ภาควิชากุมารเวชศาสตร์ โรงพยาบาลรามาธิบดี มหาวิทยาลัยมหิดล ล่าสุดได้เผยข้อมูลเกี่ยวกับโรคหายากมีอยู่จริง อย่างโรค“โกเช่ร์” เพื่อสร้างการรับรู้ข้อมูลของโรค นำมาสู่การวินิจฉัยและเข้าถึงการรักษาอย่างรวดเร็ว โดยเฉพาะพ่อแม่ที่มีพาหะของโรคทั้งคู่ พร้อมให้ความรู้พ่อแม่รับมือลูกป่วยโรคหายาก ที่ต้องเปลี่ยนแพทย์ในการรักษาบ่อย และมีค่าใช้จ่ายสูงต่อปี และพร้อมผลักดันโรคเข้าสู่ระบบหลักประกันสุขภาพแห่งชาติ ในระบบสาธารณสุขไทย และทำให้ผู้ป่วยหลายชีวิตมีคุณภาพชีวิตที่ดีขึ้น

ศ.พญ. ดวงฤดี ให้ข้อมูลว่า “ร่างกายผิดปกติ เจ็บไข้ได้ป่วยแบบหาสาเหตุไม่ได้ ท้องโต ตับโต ม้ามโต เลือดออกง่าย กระดูกหักง่าย ฯลฯ ทุกอาการที่ว่ามาถ้าทำความเข้าใจไม่มากพอ หลายคนอาจจะตีความไปว่าเป็นโรคกรรมเก่า แต่จริงๆแล้วอาจมาจากโรคทางพันธุกรรมที่ชื่อว่า “โรคโกเช่ร์” หนึ่งในโรคหายากที่หลายคนอาจจะไม่คุ้นชื่อ แต่สามารถรักษาให้อาการดีขึ้น เพื่อคุณภาพชีวิตที่ดีขึ้นได้
ทั้งนี้โรคหายากถือเป็นกลุ่มโรคที่พบได้น้อยแต่มีอยู่จริง ในประเทศไทยมีผู้ป่วยกว่า 3.5 ล้านคน จากโรคหายากกว่า 7,000 โรคที่มีอยู่บนโลกใบนี้ ประกอบกับอุบัติการณ์ผู้ป่วยโรคโกเช่รพบได้ราว 1 คนต่อ 100,000 คนเท่านั้น หลายคนจึงไม่รู้จักและไม่เคยได้ยินชื่อโรคนี้มาก่อน แต่จากรายงานต่างประเทศพบว่าผู้ป่วยโรคโกเช่ร์ จะต้องเปลี่ยนแพทย์ไปเรื่อย ๆ โดยเฉลี่ยถึง 7 คน และใช้ระยะเวลานานเป็น 10 ปี กว่าจะวินิจฉัยโรคพบ และมีค่าใช้จ่ายในการรักษาก็สูง อยู่ที่ประมาณ 2 ล้านบาทต่อปี”

สำหรับโรคนี้ซ่อนตัวในหน่วยพันธุกรรมตั้งแต่กำเนิด มักเกิดกับผู้ที่มีพ่อและแม่ที่เป็นพาหะทั้งคู่ โดยไม่ทราบว่าตนเป็นพาหะ เนื่องจากพาหะไม่แสดงอาการ แต่สามารถถ่ายทอดยีนผิดปกติไปยังลูกได้ ทำให้มีโอกาสมีลูกเป็นโรคได้ร้อยละ 25 ของทุกการตั้งครรภ์ โดยอาการของโรค โกเช่ร์ สามารถสังเกตได้จากรูปร่างที่เริ่มผิดปกติ และแสดงอาการเจ็บป่วย เช่น ท้องโตเพราะตับ ม้ามโต มีอาการทางระบบเลือด เกิดภาวะซีด เกล็ดเลือดต่ำ ทำให้เป็นแผลฟกช้ำและเลือดออกง่ายกว่าปกติ อาการปวดกระดูก มีภาวะกระดูกบาง กระดูกหักง่าย ผู้ป่วยเด็กบางรายอาจมีปัญหาเรื่องการเจริญเติบโตช้า มีความผิดปกติทางพัฒนาการด้านสติปัญญา มีอาการลมชัก เป็นต้น ปัจจุบันการรักษาโรคโกเช่ร์ในประเทศไทย มีอยู่ 2 วิธีซึ่งมีประสิทธิภาพ คือการใช้ยาเอนไซม์ทดแทนเป็นหลัก และการใช้ยาเอนไซม์ทดแทนแล้วปลูกถ่ายไขกระดูกร่วมด้วย”
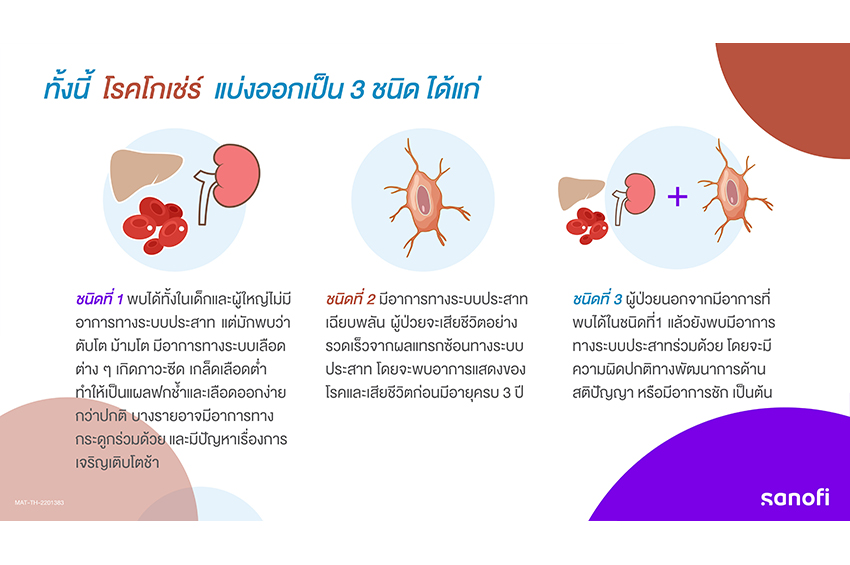
โรคโกเช่ร์ แบ่งออกเป็น 3 ชนิด ได้แก่
ชนิดที่ 1 พบได้ทั้งในเด็กและผู้ใหญ่ไม่มีอาการทางระบบประสาท แต่มักพบว่า ตับโต ม้ามโต มีอาการทางระบบเลือดต่าง ๆ เกิดภาวะซีด เกล็ดเลือดต่ำ ทำให้เป็นแผลฟกช้ำและเลือดออกง่ายกว่าปกติ บางรายอาจมีอาการทางกระดูกร่วมด้วย และมีปัญหาเรื่องการเจริญเติบโตช้า
ชนิดที่ 2 มีอาการทางระบบประสาทเฉียบพลัน ผู้ป่วยจะเสียชีวิตอย่างรวดเร็วจากผลแทรกซ้อนทางระบบประสาท โดยจะพบอาการแสดงของโรคและเสียชีวิตก่อนมีอายุครบ 3 ปี
ชนิดที่ 3 ผู้ป่วยนอกจากมีอาการที่พบได้ในชนิดที่1 แล้วยังพบมีอาการทางระบบประสาทร่วมด้วย โดยจะมีความผิดปกติทางพัฒนาการด้านสติปัญญา หรือมีอาการชัก เป็นต้น
สิ่งสำคัญในการส่งต่อความช่วยเหลือให้แก่ผู้ป่วยคือ การสร้างการรับรู้ถึงการมีอยู่ของโรค การวินิจฉัยและการเข้าถึงการรักษาอย่างรวดเร็ว ตลอดระยะเวลากว่า 20 ปีที่ซาโนฟี่ ประเทศไทย ในฐานะบริษัทชั้นนำด้านสุขภาพระดับโลก พร้อมด้วยสมาคมพันธุศาสตร์แห่งประเทศไทย สมาคมเวชพันธุศาสตร์และจีโนมิกส์ทางการแพทย์ (สวพจ.) สมาคมโลหิตวิทยาแห่งประเทศไทย มูลนิธิเพื่อผู้ป่วยโรคหายาก มูลนิธิโรคพันธุกรรมแอลเอสดี มุ่งมั่นที่จะร่วมเป็นส่วนหนึ่งในการสร้างความเปลี่ยนแปลง ให้แก่โรคหายากและยืนหยัดและอยู่เคียงข้างกับกลุ่มผู้ป่วยโรคโกเช่ร์ สร้างการตระหนักรู้ และขับเคลื่อนให้เกิดการเข้าถึงการรักษาโรคโกเช่ร์ ส่งผลให้โรคโกเช่ร์เป็นโรคพันธุกรรมเมแทบอลิกกลุ่มแรก ที่สามารถเข้าสู่ระบบหลักประกันสุขภาพแห่งชาติ ในระบบสาธารณสุขไทย และทำให้ผู้ป่วยหลายชีวิตมีคุณภาพชีวิตที่ดีขึ้น สามารถใช้ชีวิตได้อย่างปกติสุข โดยจะไม่มีใครถูกทิ้งไว้ข้างหลังอีกต่อไป